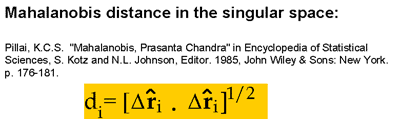|
|
|







|
Understanding the recognition of protein structural classes by amino acid composition(.pdf) |
| Ivet Bahar 1 2, Ali Rana Atilgan 2, Robert L. Jernigan 1 *, Burak Erman 2 |
| 1Molecular
Structure Section, Laboratory of Experimental and Computational
Biology, Division of Basic Sciences, National Cancer Institute,
National Institutes of Health, MSC 5677, Bethesda, Maryland 2Polymer Research Center, Bogazici University, and TUBITAK Advanced Polymeric Materials Research Center, Bebek 80815, Istanbul, Turkey |
Keywords:
non-bonded contacts; coordination of amino acids; Kirchhoff matrices; lattice models; singular value decomposition; secondary structure content prediction; contact patterns
Abstract:
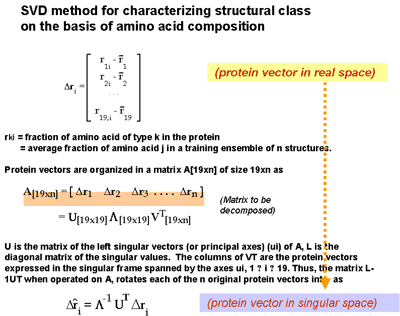
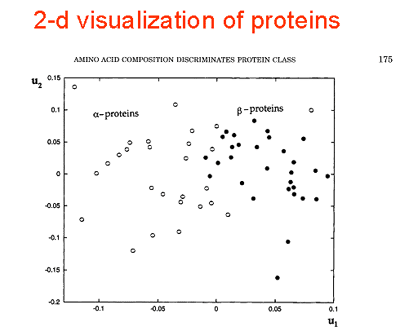
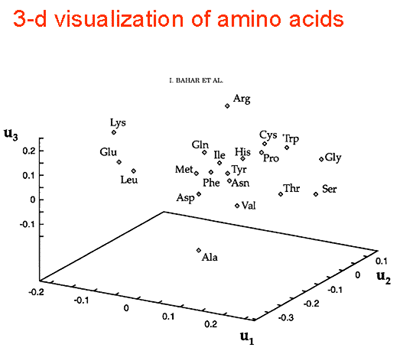
| Knowledge of amino acid composition, alone, is verified here to be sufficient for recognizing the structural class, a, b, a+b, or a/b of a given protein with an accuracy of 81%. This is supported by results from exhaustive enumerations of all conformations for all sequences of simple, compact lattice models consisting of two types (hydrophobic and polar) of residues. Different compositions exhibit strong affinities for certain folds. Within the limits of validity of the lattice models, two factors appear to determine the choice of particular folds: 1) the coordination numbers of individual sites and 2) the size and geometry of non-bonded clusters. These two properties, collectively termed the distribution of non-bonded contacts, are quantitatively assessed by an eigenvalue analysis of the so-called Kirchhoff or adjacency matrices obtained by considering the non-bonded interactions on a lattice. The analysis permits the identification of conformations that possess the same distribution of non-bonded contacts. Furthermore, some distributions of non-bonded contacts are favored entropically, due to their high degeneracies. Thus, a competition between enthalpic and entropic effects is effective in determining the choice of a distribution for a given composition. Based on these findings, an analysis of non-bonded contacts in protein structures was made. The analysis shows that proteins belonging to the four distinct folding classes exhibit significant differences in their distributions of non-bonded contacts, which more directly explains the success in predicting structural class from amino acid composition. Proteins 29:172-185, 1997. Published 1997 Wiley-Liss, Inc. |
Received: 10 February 1997; Accepted: 6 June 1997
Funding Agency: NATO Collaborative Research Grant Project; Grant
Number: CRG951240
Funding Agency: Bogazici University Research Funds Project; Grant
Number: 96A0430
|
|
|
|
|
|