Coarse Grained Simulations
Why Coarse-Grained Simulations?
To characterize protein conformational characteristics at time and
length scales that can not be reached by conventional fully atomistic MD
simulations.
The motions of nanoseconds to milliseconds time scale include:
- the correlated fluctuations between residues
- the structural changes and/or spatial reorganization of secondary
structure units
- the cooperative changes in tertiary contacts
- the larger scale motions like domain movements
The coarse-grained simulations enable exploring such collective
motions at the expense of accuracy. However, such simulations can become
efficient and physically sensible with the use of semi-empirical
potentials derived from Protein Databank Structures.
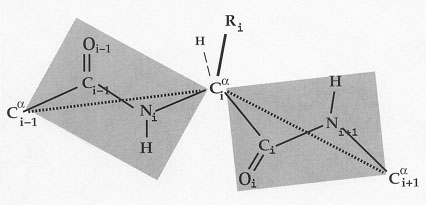
Figure I.4.6.
Virtual bond model representation of the protein backbone. Dotted lines
are the virtual bonds connecting successive
a-carbons. This representation
takes advantage of the planarity of the three successive backbone bonds,
Cai-1-Ci,
Ci-Ni
and Ni-Cai
corresponding to each amino acid.
DATA File for Long Range and Short Range Interactions
Low-resolution Model
The atomic representation is reduced to a low-resolution model where
each residue is represented by two interaction sites; one at the alpha
carbon atom and the other at the side chain centroid.
The backbone is represented by the virtual bond model originally
proposed by Flory and collaborators.The backbone of a protein of n
residues consists of n-1 virtual bonds connecting the successive
alpha carbon atoms. The amino acid side chains are represented by a
single interaction site each, selected on the basis of specific
properties.of the amino acid.
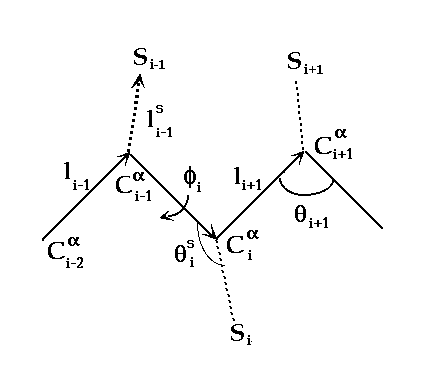
The virtual bond model where two points per residue are
considered:
the alpha carbon (Ca) as the backbone site Ci, and
the interaction site of the side chain, Si.li is
the virtual bond connecting Ci and Ci-1, phi (f) is the
torsion angle of bond l and theta (q) is the bond angle between bonds i
and i+1.
A
fortran code to calculate bond lengths, bond angles and dihedral angles
The energy of the conformation can be found from the
additive contributions of two interaction potentials:
-
Short-range: between covalently bonded first or second
neighboring units along the chain sequence
-
Long-range: between non-bonded residues that are close
in space but distant along the sequence
DATA File for Long Range and Short Range Interactions
Dynamic Monte Carlo Method
The low-resolution model of the protein structure can be
allowed to move by a dynamic Monte Carlo method.
The simple algorithm is as follows:
- calculate the energy of the conformation
- choose randomly a backbone or sidechain interaction site
- perturb the coordinates of the chosen site
- calculate the energy of the new conformation
- accept or reject the move according to Metropolis criterion
- repeat the procedure to obtain a trajectory for analysis
What can be obtained from the analysis of the simulations?
- prediction of crystallographic temperature factors
- correlation between fluctuations of units
- correlation between rotations of virtual backbone bonds
- local flexibility from conformational autocorrelations
- prediction of H/D exchange behavior
- prediction of order parameters obtained from NMR relaxation data
Related References
"Efficient Characterization of Collective Motions and Interresidue Correlations in Proteins by Low-Resolution Simulations
(.pdf)"
I. Bahar, B. Erman, T. Haliloglu & R. L. Jernigan, Biochemistry, 36,
13512-13523 (1997)
"Coarse-grained Simulations of Conformational
Dynamics of Proteins: Application to Apomyoglobin (.pdf)"
T. Haliloglu & I. Bahar, Proteins, 31, 271-281 (1998)
"Coarse-Grained Simulations of Conformational
Dynamics of Proteins"
T. Haliloglu, Theoretical and Computational Polymer Science,
9/3-4, 255-260 (1999)
"Characterization of Internal Motions of Escherichia
coli Ribonuclease H by Monte Carlo Simulation"
T. Haliloglu, Proteins, 34, 533-539 (1999)
"Conformational Dynamics of Chymotrypsin Inhibitor 2
by Coarse-Grained Simulations"
N. Kurt & T. Haliloglu, Proteins, 37, 454-464 (1999)
"Conformational Dynamics of Cytochrome c by
Coarse-Grained Simulations"
T. Haliloglu, Polymer Preprints, 81, 155 (1999).
Semi-empirical Potentials
"Structure-Derived Potentials and Protein
Simulations (.pdf)"
R. L. Jernigan & I. Bahar Current Opinion in Structural Biology
6,195-209, 1996.
"Inter-residue Potentials in Globular Proteins and
the Dominance of Highly Specific Hydrophilic Interactions at Close;
Separation"
I. Bahar & R. L. Jernigan, J. Mol. Biol., 266, 195-214
(1997)
"Short-range Conformational Energies, Secondary
Structure Propensities, and Recognition of Correct Sequence-Structure
Matches (.pdf)"
I. Bahar, M. Kaplan & R. L. Jernigan, Proteins, 29,
292-308 (1997)
"Coordination Geometry of Non-bonded Residues in
Globular Proteins"
I. Bahar & R. L. Jernigan, Folding & Design, 1
357-370 (1996)
"Empirical solvent-mediated potentials hold for both
intramolecular and inter-molecular inter-residue interactions"
O. Keskin, I. Bahar, A. Badredtinov, O. Ptitsyn & R. L. Jernigan Protein
Science 7, 2578-2586, 1998.
|






